
酒石酸托特罗定缓释片
Tolterodine Tartrate Sustained-Release Tablets
Jiu Shi Suan Tuo Te Luo Ding Huan Shi Pian
本品活性成份为酒石酸托特罗定。
化学名称:(R)-N, N-二异丙基-3-(2 -羟基-5 -甲基苯基)-3 -苯丙胺L (+)-酒石酸盐
化学结构式: 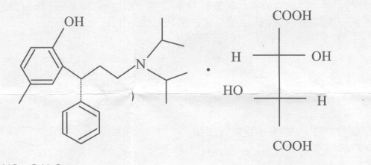
分子式:C22H31NO·C4H6O6
分子量:475.57
本品为薄膜衣片,除去包衣后显白色或类白色。
本品适用于因膀胱过度活动引起的尿频、尿急和/或急迫性尿失禁症状的治疗。
4mg
口服,推荐剂量为以一次一片(4mg),一日一次。
本品的副作用一般可以耐受,停药后即可消失。本品具有轻、中度抗胆碱能作用,可能会产生口干、消化不良和泪液减少。

下述状况禁用:
1.尿潴留、胃潴留、未经控制的窄角型青光眼。
2.已证实对本品任何成分有过敏反应。
3.重症肌无力、严重的溃疡性结肠炎、中毒性巨结肠。
1.本品为缓释片,需吞服,勿嚼碎;如需减少剂量,也可沿片面中心线完全分开,取半片服用。
2.服用本品可能引起视力模糊,用药期间驾驶车辆、开动机器和进行危险作业者应当注意。
3.肝功能明显低下的患者,遵医嘱服用。
4.肾脏功能低下的患者,自主性神经疾病患者、裂孔疝患者慎用本品。
5.由于尿潴留的风险,本品慎用于膀胱出口梗阻的病人;由于胃潴留的风险,也慎用于患胃肠道梗阻性疾病,如幽门狭窄的患者。
孕妇慎用本品。哺乳期间服用本品应停止哺乳。
妊娠小鼠给予托特罗定20mg/kg/d (相当于人类用量的14倍),未见异常和畸型。当给予妊娠小鼠30 - 40mg/kg/d的剂量时,小鼠出现死胎、胎儿体重降低,且胎鼠的异常事件的发生率增多(腭裂、异常趾、腹腔出血以及各种骨骼异常, 主要是骨化程度降低),这种剂量下的AUC值约为人类服用托特罗定时的20 - 25倍。家兔皮下给予托特罗定0.8mg/kg/d, 所得的AUC为100ug.h/L,剂量约为人类的3倍,该剂量不会引起胚胎毒性或致畸。因尚无怀孕妇女使用托特罗定缓释 片的研究情况,所以,仅当在明确了托特罗定对母亲的潜在益处大于对胎儿的潜在危险时才能在孕期使用托特罗定。
托特罗定片可从小鼠的乳汁中排泄。雌鼠在哺乳期给予托特罗定20mg/kg/d,其喂养的幼鼠体重自然增加轻微 降低,但在成熟期,体重会逐渐恢复。目前尚不清楚托特罗定是否从人类的乳汁中排泄。因此,在哺乳期不 应使用托特罗定缓释片,如需使用则应停止哺乳。
儿童应用托特罗定的安全有效性尚未建立。
老年患者与年轻患者使用托特罗定的安全性并非完全不同。
据文献报道,在一项I期临床多次给药研究中,服用托特罗定普通制剂4mg (2mg,Bid)后,老年(64-80岁)和年轻(<40岁) 健康志愿者体内的托特罗定和5-羟甲基代谢物血清药物浓度相近。另一项1期研究中,口服托特罗定普通剂2mg或4mg( 1或2mg,Bid)后,老年健康志愿者体内的托特罗定和5-羟甲基代谢物血清浓度分别比年轻健康志愿者高出20和50%。 然而在另一项有对照的12周三期临床试验中,该药物的安全性在老年患者和年轻患者之间并无不同。因此,不推荐老年患者在使用托特罗定时进行剂量调整。
与其他具有抗胆碱作用的药物合并给药时可增强治疗作用但也增强不良反应。反之毒葷碱受体激动剂可降低本品的疗效。 其他药物因需细胞色素P450 2D6 (CYP2D6)或CYP3A4进行代谢或能抑制细胞色素活性,所以可能与本品发生药代动 力学上的相互作用。如与氟西汀(氟西汀是CYP2D6的强抑制剂,氟西汀被代谢成去甲氟西汀是CYP3A4的抑制剂)合 并给药可轻度增加非结合型托特罗定及其5-羟甲基代谢物量,但这并不引起明显临床意义的相互作用。如要合并使用较 强作用的CYP3A4抑制剂如大环内酯类抗生素(红霉素和克拉霉素)、抗真菌药(酮康唑、咪康唑、伊曲康唑),应十分谨慎。 临床研究显示本品与华法林或口服避孕药(左炔诺孕酮/炔雌醇)合并给药无相互作用。
临床研究结果也未显示本品抑制了 CYP2D6、2C19、3A4或1A2的活性。
给志愿者的最大剂量是单剂给予酒石酸托特罗定12.8mg,其最严重的不良反毕是调节失调和排尿困难。在此本品过量 的事件中用洗胃和给予活性炭进行治疗。
对症状的治疗如下:
严重的中枢抗胆碱能作用(如幻觉,严重的兴奋):用毒扁豆碱治疗。
抽搐或显著的兴奋:用安定类(benzodiazepines)药物治疗。
呼吸功能不全:用人工呼吸进行治疗。
心动过速:用β阻滞剂进行治疗。
尿闭:用插导尿管进行治疗。
散瞳:用匹鲁卡品滴眼或将病人置于暗室中。
药理作用:本品用于缓解膀胱过度活动所致的尿频、尿急和急迫性尿失禁症状,为竞争性M胆碱受体阻滞剂。动物试验结果提示本品对膀胱的选择性高于唾液腺,但尚未得到临床的证实。
本品口服后经肝脏代谢成起主要药理作用的活性代谢产物5-羟甲基衍生物,其抗胆碱活性与本品相近。两者对M胆碱受体均具有高选择性,对其他神经递质的受体和潜在的细胞靶点(如钙通道)的作用或亲和力很弱。
据文献资料:托特罗定口服后可迅速地吸收,吸收率大于77%。食物的摄入,年龄和性别的差别不需调整剂量。托特罗 定口服1〜4mg,最大血药浓度(Cmax)和药时曲线下面积(AUC)与剂量呈线性关系。口服托特罗定2mg后,2.5小 时左右达到峰值血药浓度,Cmax为2.5ug / L,AUC为11.8ug/ L·h。5-羟甲基活性代谢物(DD01)的血药浓度与本品原形极其相似,为2.2ug / L,AUC为12.1ug / L • h。
根据未结合型酒石酸托特罗定的血药浓度,口服酒石酸托特罗定缓释片4mg,每天1次的AUC等同于口服酒石酸托特罗 定片2mg,每天2次的AUC。缓释片Cmax降低,Tmax延长。血清酒石酸托特罗定最高浓度出现在给药后2-6小时。
托特罗定与血浆蛋白结合率较高,游离的托特罗定的浓度平均为3.7%±0.13%。DD01与血浆蛋白结合率不高,游离DD01的浓度平均为36%±4.0%。托特罗定与其代谢物DD01在血液与血浆的比值分别为0.6和0.8。静脉注射托特罗定 1.28mg的分布容积为113±26.7L。
托特罗定口服给药后经历肝脏广泛的首过效应,主要的代谢途径涉及5-甲基位的氧化.此过程由CYP2D6参与,形成具有药理活性的5-羟甲基代谢物(DD01)。进一步的代谢将产生5-羧酸和N-脱烷基 5-羧酸代谢物,在尿中回收的这2种代谢物分别为51%±14%和29%±6.3%。托特罗定消除半衰期(t1/2)为2~3h, DD01的t1/2为3~4h。
给健康志愿者口服14C标记的托特罗定片5mg后,尿、粪排泄率分别为77%、17%,原形托特罗定的排泄率不到给药量的1%,5%~14%以活性DD01回收。在给药后24h内大多数放射活性物自人体内排泄。
密封,常温(10-30℃)保存。
铝塑包装.4片/板/盒,7片/板/盒,10片/板/盒,14片/板/盒。
24个月
YBH04832007
国药准字H20070272
FDA妊娠分级:C















