
尼洛替尼胶囊
达希纳® Tasigna®
Nilotinib Capsules
Ni Luo Ti Ni Jiao Nang
警示语:QT间期延长和猝死
已经有接受尼洛替尼治疗的患者猝死的报告。尼洛替尼不可用于低血钾、低血镁或长QT综合征的患者。在使用尼洛替尼以前必须纠正低钾和低镁,并定期进行监测。避免合用已知的可延长QT间期的药物和CYP3A4的强效抑制剂。在给药前2小时和给药后1小时避免进食。有肝功能损害的患者建议减量。在开始给药前、开始给药后7天以及之后时间里定期进行ECG检查以监测QTc,并且在任何时候进行剂量调整时也应如此。
本品活性成份为尼洛替尼。
化学名称:4-甲基-N-[3-(4-甲基l-1氢-咪唑-1-基)-5-(三氟甲基)苯基]-3-[(4-吡啶-3-基嘧啶-2-基)氨基] 苯甲酰胺单盐酸盐一水合物
化学结构式: 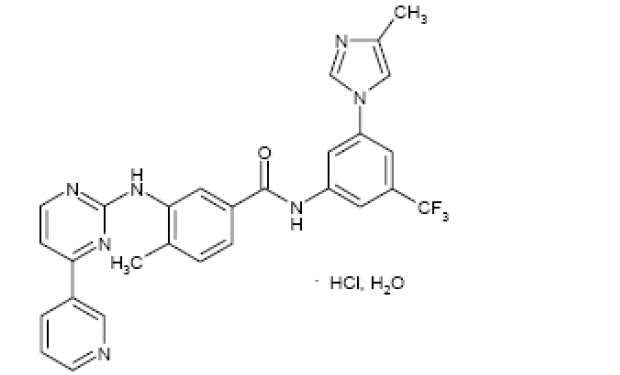
分子式:C28H22F3N7O·HCl·H2O
分子量:583.99
本品内容物为白色至黄色粉末。
用于治疗新诊断的费城染色体阳性的慢性髓性白血病(Ph+CML)慢性期成人患者。
用于对既往治疗(包括伊马替尼)耐药或不耐受的费城染色体阳性的慢性髓性白血病(Ph+CML)慢性期或加速期成人患者。
每粒含有200 mg尼洛替尼,150mg尼洛替尼。
本品的初始治疗应该在对CML患者有治疗经验的医师指导下进行。
新诊断的 Ph+ CML 慢性期
本品的推荐剂量为 300 mg 每日两次口服。只要患者持续受益,本品治疗应持续进行。
已获得持续深度分子学反应(MR4.5)的新诊断为 Ph+ CML-CP 的患者用量。
既往接受本品300mg每日两次治疗至少3年的Ph+ CML-CP患者,如果在终止治疗之前的深度分子学反应持续至少1年,可考虑终止治疗。应该由具有CML患者治疗经验的医师来启动本品的停药程序(见【注意事项】、【临床试验】)。
在1年内必须每月监测一次符合停药条件的患者的BCR-ABL转录本水平及其全血细胞计数和分类,随后在第2年每6周监测一次,其后则每12周监测一次。
丧失主要分子学反应(MMR)的患者必须在得知出现丧失反应后4周内重新开始治疗。应该以300 mg每日两次的剂量重新开始治疗,或者如果患者在终止治疗之前已下调治疗剂量,则以400 mg每日一次的减低剂量重新开始本品治疗。每月监测一次重新使用本品的患者的BCR-ABL转录本水平,直到重新获得主要分子学反应(见【注意事项】、【临床试验】)。
耐药或不耐受的 Ph+ CML 慢性期或加速期
本品的推荐剂量为 400mg 每日两次口服。只要患者持续受益,本品治疗应持续进行。
对伊马替尼不耐受的定义是:尽管采用了最佳支持治疗,在任何剂量和/或治疗期间,患者仍由于 3 或 4 级不良事件的持续存在而中止伊马替尼治疗;或者,尽管采用了最佳支持治疗,与伊马替尼治疗相关的 2 级不良事件仍持续时间≥1 个月,或反复发生超过 3 次,不论是否剂量减少或中止治疗。
推荐剂量为每日 2 次,间隔约 12 小时,不得与食物一起服用。在服药前至少2 小时以及服药后至少 1 小时内不得进食。
胶囊应用水完整吞服,不应咀嚼或吮吸,不应打开胶囊。手接触胶囊后应立即清洗。小心不要吸入胶囊中的任何粉末(比如胶囊损坏),也不要让药粉接触皮肤或粘膜。如果发生皮肤接触,用肥皂和水清洗局部。如果眼睛接触了药粉,用水冲洗。如果胶囊中的药粉撒出,应该用手套和可弃去的湿毛巾擦去,置于密封的容器中正确丢弃。
对于不能吞咽胶囊的患者,可以把胶囊的内容物与一茶匙的苹果酱混合在一起,混匀后应立即服用。苹果酱不能超过一茶匙,同时不能食用除了苹果酱以外的其他食物。(见【注意事项】)。
应遵医嘱治疗。但如果错过给药,患者不得另外补充剂量,而是按照处方服用下一次剂量。
如果临床需要,本品可与造血生长因子如EPO或G-CSF联合使用,也可与羟基脲或阿那格雷联用。
既往伊马替尼治疗后接受本品治疗且已取得持续深度分子学反应(MR4.5)的 Ph+ CML-CP 患者的用量
既往接受本品治疗至少3年的Ph+ CML-CP患者,如果在终止治疗之前的深度分子学反应持续至少1年,可考虑终止治疗。应该由具有CML患者治疗经验的医师来启动尼洛替尼的停药程序(见【注意事项】、【临床试验】)。
在1年内必须每月监测一次符合停药条件的患者的BCR-ABL转录本水平及其全血细胞计数和分类,随后在第2年每6周监测一次,其后则每12周监测一次。
确认丧失MR4.0(相隔至少4周的两次连续测定显示丧失MR4.0)或丧失主要分子学反应(MMR)的患者必须在得知丧失反应后4周内重新开始治疗。应该以300mg或400mg每日两次的剂量重新开始本品治疗。每月监测一次重新使用本品治疗的患者的BCR-ABL转录本水平,直到重新取得之前的主要分子学反应或MR4.0(见【注意事项】、【临床试验】)。
监测建议和剂量调整
对接受本品的患者,应该根据医生的判断进行一定频率的实验室检查。
在最初的 2 个月,应每隔 2 周做一次全血细胞计数,之后可每个月检测一次。应定期检查生化。
应定期或当治疗发生改变时对本品治疗在 Ph+ CML 患者中的应答情况进行监测,以确定疗效欠佳、对治疗无应答、患者依从性不佳或可能的药物相互作用。监测结果应指导恰当管理 CML。
建议在开始本品治疗前进行基线心电图检查,在7天后和有任何临床指征时再进行心电图检查。在本品给药前必须纠正低钾血症或低镁血症,在治疗期间必须对钾和镁的血药浓度进行定期检测,尤其是对于有电解质异常风险的患者而言更应该严密监测(见【注意事项】)。
已有本品治疗中的总血清胆固醇水平升高报告。在开始本品治疗前应进行血脂评估,并在治疗过程中有临床指征时进行评估。
已有本品治疗中的血糖水平升高报告(见【注意事项】)。在开始本品治疗前应进行血糖评估,并在治疗过程中进行监测。
由于可能发生溶瘤综合征(TLS),建议在开始本品治疗前,纠正有临床表现的脱水并治疗高尿酸血症。(见【不良反应】)。
如果心电图显示QTc> 480毫秒,则应停止服用本品,及时检测血清钾和镁,如果血清钾和镁低于正常值低限,则应补充使之达到正常范围,并必须检查合并用药的情况;如果QTcF恢复到<450毫秒,并与基线值相差不超过20毫秒,则可在2周内恢复本品先前的剂量;如果2周后,QTcF在450毫秒和480毫秒之间,则应降低本品剂量至每日一400mg;如果降低剂量至每日400mg后,QTcF仍> 480毫秒,则应停止使用本品。任何一次的剂量调整,均应在7天后复查心电图。
由于与潜在白血病不相关的血液学毒性(中性粒细胞减少、血小板减少),有时候可能需要暂时停止本品和/或降低其剂量,见表 1。
表1 由于中性粒细胞减少和血小板减少而进行的剂量调整

如果出现有显著临床意义的中度或严重的非血液学毒性,应该中止服药;一旦毒性缓解,可以恢复每日一次,每次400mg的剂量。如果临床上适合,应考虑将剂量逐步恢复至 300mg(新诊断的 Ph+ CML 慢性期)或 400mg(耐药或不耐受的 Ph+CML 慢性期或加速期),每日两次。
血清脂肪酶升高:如果出现3~4级血清脂肪酶升高,剂量应降低至每日一次,每次400mg或中止给药。应每月监测血清脂肪酶或遵医嘱。
胆红素和肝转氨酶升高:如果出现3~4级胆红素升高,剂量应降低至每日一次,每次400mg或中止给药。应每月监测胆红素和转氨酶或遵医嘱。
同时使用强效CYP3A4抑制剂
避免同时使用强效CYP3A4抑制剂(例如,酮康唑,伊曲康唑,克拉霉素,阿他那韦,茚地那韦,奈法唑酮,那非那韦,利托那韦,沙奎那韦,泰利霉素,伏立康唑等)。葡萄柚产品也可增加尼洛替尼的血药浓度,应避免食用。如果需要应用任何上述药物治疗,建议中止本品治疗。如果患者必须同时使用强效CYP3A4抑制剂,根据药物代谢动力学研究,剂量应减少至300 mg每日一次。然而,在应用强效CPY3A4抑制剂的患者中尚无该剂量调整的临床数据。如果中断应用强效抑制剂,在本品剂量上调至适用剂量前应经过一个洗脱期。对不能避免使用强效CYP3A4抑制剂的患者,应对QT间期延长进行密切的监测。
同时使用强效 CYP3A4 诱导剂
避免同时应用强效 CYP3A4 诱导剂(例如,地塞米松,苯妥英,卡马西平,利福平,利福布汀,利福喷汀,苯巴比妥)。患者还应避免服用贯叶连翘,因为这些药物可能会降低本品的浓度。根据本品的非线性药代动力学特征,当与这些药物同时给药时,增加本品的剂量不可能补偿药物暴露的降低。
特殊剂量推荐
儿童和青少年
尚无在儿童或青少年中进行的临床研究。所以不推荐用于治疗小于18岁的患者。
老年患者
临床研究(新诊断的 Ph+CML 慢性期及耐药或不耐受的 Ph+CML 慢性期或加速期)中大约有 12%-30%的受试者为 65 岁或以上患者。与 18-65 岁年龄组的成年受试者相比较,在≥65 岁的患者中没有观察到安全性及有效性方面的差异。
对超过65岁的患者,不需要进行特殊的剂量调整。
肾损害患者
尚未在肾损害患者中进行的临床研究。
本品及其代谢产物只有少部分经肾排泄,所以预计肾损害患者并不会出现总体清除率的降低。对肾损害患者,不需要进行剂量调整。
肝损害患者
对转氨酶超过正常值2.5倍或胆红素升高超过正常值1.5倍的肝损害患者,不推荐本品治疗。(见【注意事项】)
如果可能,考虑替代疗法;若必须使用尼洛替尼胶囊,伴有肝损害患者应考虑剂量减少,见表2。

安全性特征摘要
尼洛替尼的安全性基于一项随机化、开放标签、活性对照剂、III 期试验中的新诊断的 Ph+ CML 慢性期患者和另一项研究中的耐药或不耐受的 Ph+ CML 慢性期和加速期患者的数据,这些数据是收载适应症的基础。还给出了来自两项尼洛替尼治疗终止研究以及来自一项在伊马替尼疗效欠佳的慢性期 Ph+ CML 患者中进行的 III 期研究的安全性信息。
新诊断的 Ph+ CML 慢性期患者
以下所报告的数据反应了一项III期随机临床研究中的本品暴露情况,该研究以新诊断的慢性期Ph+ CML患者为对象,研究中这些患者接受推荐的300 mg每日两次剂量(n=279)。治疗中位时间为60.5个月(范围0.1 -70.8个月)。
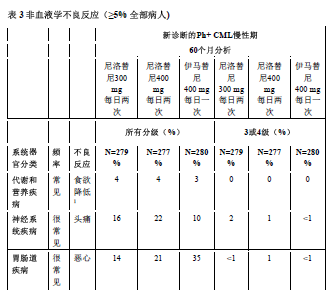
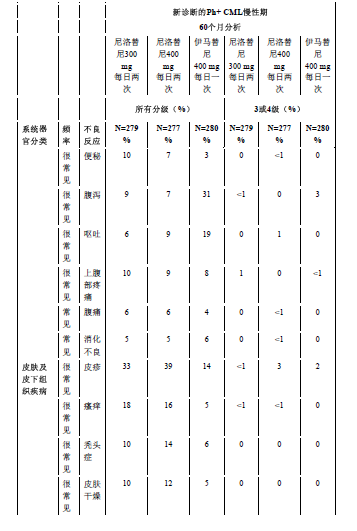
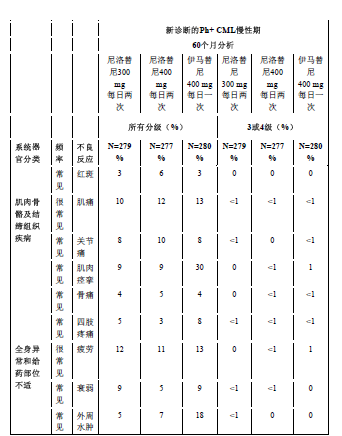
最常见报告的(≥10%)非血液学药品不良反应为皮疹、瘙痒、头痛、恶心、疲劳、脱发、肌肉疼痛和上腹部疼痛。大多数药品不良反应为轻度或中度(1级或2级)。据观察,不常见(< 10% 且≥ 5%)不良反应有:便秘、腹泻、皮肤干燥、肌肉痉挛、关节痛、腹部疼痛、周围水肿、呕吐和虚弱,这些不良反应都为轻度至中度,易控制且一般都不需要降低剂量。
在接受本品300mg每日两次治疗的患者中,不论因果关系如何,分别有2%和<1%的患者出现胸腔和心包积液。3%的患者出现胃肠道出血。在接受本品300mg每日两次的推荐剂量组中观察到,稳态下的一段时间内的QTcF间期平均值与基线时相比发生变化为6毫秒。在本品400mg每日两次组及伊马替尼400mg每日一次组中,稳态下的一段时间内的QTcF间期平均值变化分别
为6毫秒和3毫秒。在任何治疗组中,无患者的绝对QTcF >500毫秒,且未观察到尖端扭转型室性心动过速。在5位患者中观察到使用研究药物时与基线时相比QTcF间期变化为60毫秒(1例在300mg每日两次治疗组,4例在400mg每日两次治疗组)。
在治疗过程中,任何治疗组中都没有患者出现LVEF<45%。同时,所有患者的LVEF与基线时相比降低程度都低于15%。
任何治疗组都没有报告有猝死。
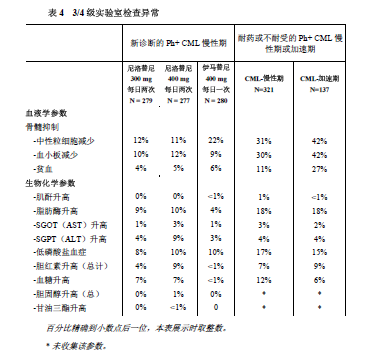
在本品300mg每日两次组,血液学药物不良反应包括骨髓抑制:血小板减少症(18%)、中性粒细胞减少(15%)、及贫血(8%)。生化不良反应包括丙氨酸氨基转移酶升高(24%),高胆红素血症(16%),天门冬氨酸氨基转移酶升高(12%),脂肪酶升高(11%),血胆红素升高(10%),高血糖症(4%),高胆固醇血症(3%)以及高甘油三酯血症(<1%)。3/4级实验室异常见表4。
观察到10%的患者由于药物不良反应导致治疗终止。
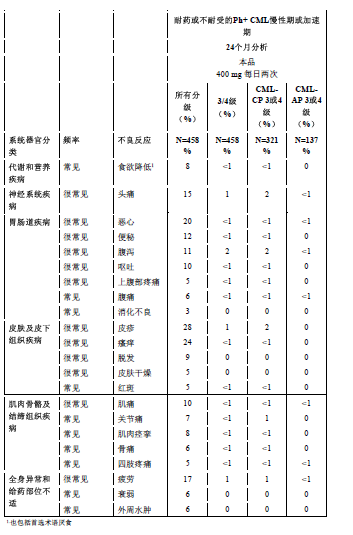
耐药或不耐受的 Ph+ CML-CP 和 CML-AP 的患者
以下数据来自于一项458例慢性髓性白血病慢性期(321例)或加速期(137例)患者接受尼洛替尼治疗的开放、多中心研究。这些患者对包括伊马替尼在内的既往至少一种治疗耐药或不耐受。不论因果关系怎样,因不良事件而导致试验终止的患者在慢性期组中为16%,在加速期组中为10%。
最常见的(在慢性髓性白血病慢性期和加速期两个群组中共≥10%)与药物相关的非血液学不良反应是皮疹、瘙痒症、恶心、疲劳、头痛、便秘、腹泻、呕吐、肌肉痛。大多数不良反应为轻度至中度。脱发、肌肉痉挛、食欲减退、关节痛、骨痛、腹痛、外周性水肿及乏力相对较少见(<10% 且≥5%),为轻度至中度(1级或2级)。
接受尼洛替尼治疗的患者中有<1%的患者出现了胸膜和心包渗出以及体液潴留并发症。有<1%的患者发生了心脏衰竭。分别有1%和<1%的患者报告了胃肠道和中枢神经系统出血。
此次研究中观察到有4例患者(<1%)的QTcF超过了500毫秒。没有观察到尖端扭转型室性心动过速发作(暂时的或持续的)。
血液学不良反应包括骨髓抑制:血小板减少症(31%)、中性粒细胞减少症(17%)、和贫血(14%)。3/4级的实验室检查异常见表格4。
该研究观察到 16%的慢性期患者和 10%的加速期患者由于药物不良反应而导致治疗终止。
伊马替尼治疗后未获得大于或等于 4.5 个对数级下降的的分子学反应的Ph+CML-CP 患者中
下文报告数据来自一项随机、开放性、III期研究,诊断为Ph+ CML-CP的成年男性和女性患者在经过2年的伊马替尼治疗之后接受了48个月的尼洛替尼400mg每日一次给药或者伊马替尼400 mg或600 mg每日一次给药治疗。随机分配至伊马替尼组的患者接受了与随机分组之前一致的伊马替尼剂量。尼洛替尼组以及伊马替尼组400 mg和600 mg组的中位暴露时间分别为47.2个月、37.0个月和26.7个月。
在尼洛替尼组至少20%的患者报告的不良反应并且比伊马替尼组患者更多报告的药物不良反应为头痛、皮疹和瘙痒。与伊马替尼组患者相比,更高比例的尼洛替尼组患者报告了导致停药的AE和需要调整剂量/中断给药的AE。尼洛替尼治疗后常报告胆红素和转氨酶升高。
至48个月的截止日期为止,已经观察到了3起治疗中死亡事件(尼洛替尼组2起、伊马替尼组1起)。3名患者在停用研究药物后28天之外死亡(尼洛替尼组1名、伊马替尼组2名)。
在第8天,在4名接受尼洛替尼治疗的患者中观察到QTc间期>450毫秒。无患者QTc间期>480毫秒。8名患者(7.9%)报告了QTc间期相对于基线延长>30毫秒。尼洛替尼组患者没有出现QTc间期延长>60 毫秒。
表3显示了在尼洛替尼的任何临床研究中至少有5%的患者报告的非血液学不良反应(不包括实验室异常)。按发生频率排序,最常见的在先,各频率组内,药物不良反应按严重性降序分布。此外,各药物不良反应相应的频率分类使用下述定义(CIOMS III):很常见(≥ 1/10)或常见(≥ 1/100,<1/10)。频率以研究中所有尼洛替尼组最高的发生率为依据,精确到百分比的小数点后一位。


来自临床研究的其它资料
以下是在尼洛替尼临床研究患者中报告的发生频率小于5%的不良反应(常见:≥ 1/100~<1/10;不常见:≥1/1000~<1>感染和侵染
常见:毛囊炎、上呼吸道感染(包括咽炎、鼻咽炎、鼻炎)
不常见:肺炎、尿路感染、胃肠炎、支气管炎、疱疹病毒感染、念珠菌病(口腔念珠菌病)
发生频率未知:脓血症、皮下脓肿、肛门脓肿、疔、足癣、乙肝病毒再激活
良性、恶性肿瘤及性质未明肿瘤
常见:皮肤乳头状瘤
发生频率未知:口腔乳头状瘤、副蛋白血症
血液和淋巴系统异常
常见:白细胞减少、嗜酸性粒细胞增多、发热性中性粒细胞减少、全血细胞减少、淋巴细胞减少
发生频率未知:血小板增多、白细胞增多
免疫系统异常
发生频率未知:超敏反应
内分泌异常
不常见:甲状腺机能亢进、甲状腺机能减退
发生频率未知:继发性甲状旁腺机能亢进、甲状腺炎
代谢和营养失衡
很常见:低磷血症(包括血磷下降)
常见:电解质失调(包括低镁、高钾、低钾、低钠、低钙、高钙、高磷酸盐)、糖尿病、高血糖、高胆固醇血症、高脂血症、高甘油三酯血症
不常见:痛风、脱水、食欲增加、血脂异常
发生频率未知:高尿酸血症、低血糖
精神异常
常见:抑郁、失眠、焦虑
发生频率未知:定向障碍、意识模糊状态、健忘、烦躁
神经系统异常
常见:头昏、周围神经病变、感觉减退、感觉异常
不常见:颅内出血、缺血性中风、短暂性脑缺血发作、脑梗死、偏头痛、意识丧失(包括晕厥)、震颤、注意力不集中、感觉过敏。
发生频率未知:脑血管意外、基底部动脉狭窄、脑水肿、视觉神经炎、嗜睡、感觉迟钝、不宁腿综合征
眼异常
常见:眼睛出血、眼窝外周水肿、眼瘙痒、结膜炎、眼干(包括干眼病)
不常见:视觉障碍、视力模糊、视敏度下降、眼睑水肿、闪光幻觉、充血(巩膜、结膜、眼部)、眼刺激、结膜出血
发生频率未知:视神经乳头水肿、复视、畏光、眼睛肿胀、眼睑炎、眼痛、脉络膜视网膜炎、过敏性结膜炎、眼表疾病
耳和迷路异常
常见:眩晕
发生频率未知:听觉损伤、耳痛、耳鸣
心血管异常
常见:心绞痛、心律不齐(包括心室传导阻滞、心脏扑动、期前收缩、心动过速、心房纤颤、心动过缓)、心悸、QT间期延长
不常见:心衰、心肌梗死、冠状动脉疾病、心杂音、心包积液、发绀
发生频率未知:心室功能异常、心包炎、射血分数降低
血管异常
常见:高血压、潮红
不常见:高血压危象、周围动脉闭塞性疾病、间歇性跛行、肢体动脉狭窄、血肿、动脉硬化
发生频率未知:出血性休克、低血压、血栓形成、周围动脉狭窄
呼吸道异常
常见:呼吸困难、劳力性呼吸困难、鼻衄、咳嗽、发声困难
不常见:肺水肿、胸膜积液、间质性肺病、胸膜疼痛、胸膜炎、咽喉疼痛、咽喉刺激
发生频率未知:肺动脉高压、喘鸣、口咽痛
消化系统异常
常见:胰腺炎、腹部不适、腹胀、消化不良、味觉障碍、胃肠胀气
不常见:胃肠道出血、黑便、口腔溃疡、胃食管返流、口腔炎、食管绞痛、口干、胃炎、牙齿过敏
发生频率未知:胃十二指肠溃疡穿孔、腹膜后出血、吐血、胃溃疡、溃疡性食管炎、不完全肠梗阻、小肠结肠炎、痔疮、食管裂孔疝、直肠出血、牙龈炎
肝胆系统异常
很常见:高胆红素血症(包括血液胆红素升高)
常见:肝功能异常
不常见:肝毒性、中毒性肝炎、黄疸
发生频率未知:胆汁淤积、肝肿大
皮肤和皮下组织异常
常见:盗汗、湿疹、荨麻疹、多汗、挫伤、痤疮、皮炎(包括过敏性、剥脱性的和痤疮样的)、皮肤干燥
不常见:剥脱性皮疹、药疹、皮肤疼痛、瘀斑、面部水肿
发生频率未知:银屑病、多形性红斑、结节性红斑、皮肤溃疡、手足口病、瘀点、光过敏、水泡、皮肤囊肿、皮脂腺增生、皮肤萎缩、皮肤褪色、皮肤剥脱、皮肤色素沉着、皮肤增生、角化过度
肌肉骨骼系统
常见:肌肉骨骼性胸痛、肌肉骨骼疼痛、背痛、颈痛、胁腹部痛、肌无力
不常见:肌肉骨骼僵硬、关节肿胀
发生频率未知:关节炎
肾和泌尿系统异常
常见:尿频
不常见:排尿困难、尿急、遗尿
发生频率未知:肾衰竭、血尿、尿失禁、色素尿
生殖系统和乳腺异常
不常见:乳腺疼痛、男子女性型乳房、勃起障碍
发生频率未知:乳腺硬化、月经过多、乳头肿胀
全身性异常
常见:胸痛(包括非心源性胸痛)、疼痛、发热、胸部不适、不适
不常见:面部水肿、重力性水肿、流感样症状、寒战、体温感觉异常(包括感觉发热、感觉冷)
发生频率未知:局部水肿
检查(3/4级实验室检查异常)
很常见:丙氨酸氨基转移酶升高,天门冬氨酸氨基转移酶升高,脂肪酶升高,脂蛋白胆固醇升高(包括极低密度脂蛋白和高密度脂蛋白),总胆固醇升高,血甘油三酯升高。
常见:血红蛋白降低、血淀粉酶升高、血碱性磷酸酶升高、γ-谷氨酰转移酶升高、肌酸磷酸激酶升高、血胰岛素升高、体重降低、体重增加、球蛋白降低
不常见:血乳酸脱氢酶升高、血尿素升高。
发生频率未知:肌钙蛋白增加、血未结合胆红素增加、血胰岛素降低、胰岛素C-肽降低、血甲状腺旁素升高
临床相关的或严重的常规血液学或生化学实验室检测值的异常,见表4。
取得持续深度分子学反应的Ph+ CML-CP患者中的治疗终止
在尝试无治疗缓解(TFR)范围内的尼洛替尼治疗终止之后,患者可能会出现相较于治疗终止之前更为频繁的肌肉骨骼症状,如肌痛、四肢疼痛、关节痛、骨痛、脊柱疼痛或肌肉骨骼疼痛。
在一项新诊断的Ph+ CML-CP患者的II期临床研究(N=190)中,有24.7%的患者在停用尼洛替尼的一年内报告了肌肉骨骼症状,而16.3%的患者在停用尼洛替尼之前应用其治疗的一年内报告了肌肉骨骼症状。
在一项尼洛替尼治疗既往接受过伊马替尼的Ph+ CML-CP患者的II期临床研究(N=126)中,有42.1%的患者在停用尼洛替尼的一年内报告了肌肉骨骼症状,而14.3%的患者在停用尼洛替尼之前应用其治疗的一年内报告了肌肉骨骼症状。
来源于自发性报告及文献病例的药物不良反应
下列不良反应通过自发性病例报告、文献病例、扩大用药项目(EAP)和全球注册试验以外的其他临床试验等上市后经验获得。因为不确定这些不良反应自愿报告的人群大小,所以不能准确地估算不良反应的发生率或者确定不良反应与尼洛替尼暴露之间的因果关系。
发生率不明:接受本品治疗的患者中有溶瘤综合征的病例报告。
♦对本品活性物质或者任何赋形剂成份过敏者禁用。
♦伴有低钾血症、低镁血症或长QT综合症的患者禁用。
骨髓抑制:本品能引起3/4级血小板减少、中性粒细胞减少和贫血。在伊马替尼耐药或不耐受的CML患者中发生频率更高,尤其是CML加速期患者。在最初的2个月,应每隔2周做一次全血细胞计数,之后可每个月检测一次,或者在有临床指征时进行。骨髓抑制一般是可逆的,可以通过暂时停用本品或降低剂量来控制。
QT间期延长:已经显示本品能延长心室复极,可通过心电图上的QT间期检测出来,呈剂量依赖性。QT间期延长能够引起尖端扭转型室性心动过速,可能引起昏厥、惊厥和/或死亡。在基线时、服药开始7天后、有临床指征时应定期做心电图,在剂量调整之后也需要做心电图。
本品禁用于低钾血症和低镁血症或长QT综合征的患者。在使用本品之前,应纠正低钾血症和低镁血症,并在治疗期间定期监测电解质。如果本品与食物同时服用(为不适当的给药方法),和/或与强效CYP3A4抑制剂和/或其他已知可潜在延长QT的药物服用时,可能会出现有临床意义的QT间期延长。因此,必须避免与食物共同服用,并应避免使用已知延长QT间期的药物和强CYP3A4抑制剂。低钾血症和低镁血症的出现可能会增加患者QT间期延长的风险。
在新诊断的Ph+CML慢性期患者的III期研究中,本品300mg每日两次剂量组中观察到,稳态下的时均QTcF间期平均值与基线时相比变化为6毫秒。在推荐的300mg每日两次剂量组中,没有患者的绝对QTcF大于480毫秒,没有观察到尖端扭转型室性心动过速事件。
在以伊马替尼耐药或不耐受的慢性期和加速期CML患者为对象的II期临床研究中,受试者接受400 mg每日两次的尼洛替尼治疗,据观察,稳态下的时均QTcF间期平均值与基线时相比变化分别为5和8毫秒。在4位患者(在总患者中所占比例<1%)中观察到QTcF间期>500毫秒。
在以健康志愿者为对象的暴露研究中,暴露量与患者中观察到的暴露量可比,时均QTcF间期平均值去除安慰剂(影响)后较基线变化为7毫秒(CI ± 4毫秒)。没有患者出现QTcF >450毫秒。此外,在试验期间没有观察到临床相关性心律失常。特别是,没有观察到尖端扭转型室性心动过速(无论短暂性或持续性)。
在具有QTc延长或存在显著QTc延长风险的患者中,要慎用本品,例如具有不可控制或临床显著性的心脏疾病患者,包括新近的心肌梗塞、充血性心力衰竭、不稳定型心绞痛或临床显著性心动过缓。
猝死:临床试验中,在接受本品治疗的具有心脏病史或显著心脏病风险的伊马替尼耐药或不耐受的慢性期或加速期的CML患者中,猝死事件较罕见(0.1至1%)。当与其他药物合用时,除潜在恶性肿瘤之外的并发症出现频率较高,室性复极化异常可能为主要因素。根据上市后的暴露量(患者- 年),估计猝死自发报告率为每患者-年0.02%。在新诊断的Ph+CML慢性期患者的III期研究中,没有猝死报告。
心血管事件
在一项新诊断的Ph+CML慢性期患者的随机、III期的尼洛替尼研究中,以及上市后报告中已报道心血管事件。临床试验的60.5个月中位治疗时间中,3/4级的心血管事件包括周围动脉闭塞性疾病(300mg和400mg每日两次剂量组发生率分别为1.4% 和 1.1%),缺血性心脏病(300mg和400mg每日两次剂量组发生率分别为2.2%和6.1%),缺血性脑血管事件(300mg和400mg每日两次剂量组发生率分别为1.1%和2.2%)。如果出现心血管事件的急性体征或症状,建议患者立即寻求治疗。应评估患者的心血管状态,在本品治疗期间根据标准治疗指南监测心血管风险因素,并积极治疗。
体液潴留
在一项新诊断的 CML 慢性期患者的 III 期研究中,已观察到严重的体液潴留,如胸腔积液、肺水肿、心包积液,该不良反应不常见(0.1-1%)。在上市后报告中也观察到类似事件。应该谨慎地研究非预期的,快速的体重增加。如在尼洛替尼用药期间有严重的体液潴留出现,应该评价其病因,并进行相应的治疗(见【用法用量】)。
乙肝病毒再激活
乙肝病毒 (HBV)慢性携带者在接受 BCR-ABL 酪氨酸激酶抑制剂(TKI)(例如尼洛替尼)之后可能发生 HBV 再激活。在某些病例中,与使用 BCR-ABL TKI 类药物有关的 HBV 再激活引发急性肝衰竭或暴发性肝炎,并从而导致肝移植或致命性结局(见【不良反应】)。
患者在开始尼洛替尼治疗之前,需检测是否存在乙肝病毒感染。当前正在使用尼洛替尼的患者需接受基线乙肝病毒病毒检测以识别出慢性乙肝病毒携带者。乙肝病毒血清学阳性的患者(包括疾病活动期的患者)及在治疗过程中检测发现乙肝病毒阳性的患者,在开始尼洛替尼治疗前应咨询肝病和乙肝治疗方面的专家。对需要尼洛替尼治疗的乙肝病毒携带者,在整个治疗期间以及治疗终止后数月应当严密监测活动性乙肝病毒感染的症状和体征。
对已取得持续深度分子学反应的Ph+CML-CP患者的特殊监测终止治疗的条件
符合条件的患者如果确认表达典型BCR-ABL转录本e13a2/b2a2或e14a2/b3a2,可考虑终止治疗。患者必须具有典型的BCR-ABL转录本,以能对BCR-ABL水平进行定量检测、对分子学反应的深度进行评估以及对终止本品治疗后可能出现的丧失分子学反应进行测定。
对终止治疗患者的监测
必须以确认测定分子学反应水平灵敏度至少MR4.5的定量诊断检测来监测符合终止治疗条件的患者的BCR-ABL转录本水平。必须在治疗终止之前和终止治疗期间评估BCR-ABL转录本水平(见【用法用量】、【临床试验】)。
丧失主要分子学反应(MMR)或确认丧失MR4.0(相隔至少4周的两次连续测量显示丧失MR4.0)将在得知丧失反应后4周内重新开始治疗。需要频繁监测BCR-ABL转录本水平以及全血细胞计数和分类,以检测可能出现的缓解丧失(见【用法用量】、【临床试验】)。
血清脂肪酶升高:使用本品会引起血清脂肪酶升高。建议慎用于有胰腺炎病史的患者。应该定期监测血清脂肪酶水平。如果脂肪酶升高伴随腹部症状,应该中断本品给药,应给予适当诊断以排除胰腺炎。
肝功能异常:使用本品可能引起胆红素、ALT/AST和碱性磷酸酶升高,应定期进行肝功能检测。
电解质异常 :使用本品可能引起低磷、低钾、高钾、低钙和低钠血症。在开始使用本品之前必须纠正电解质异常,治疗过程中应定期监测电解质。
药物相互作用 :避免使用CYP 3A4强诱导剂或延长QT间期的药物。如果患者必须使用这样的药物治疗,应该考虑停止本品的服用 ;如果不能停止本品的治疗,并需要同时服用上述药物时,应密切监测QT间期。见[药物相互作用]。
本品与潜在CYP3A4诱导剂类药物合并使用可能会使尼洛替尼暴露降低至有临床相关性的程度。因此,在接受本品治疗的患者中,合并用药应该选择CYP3A4诱导潜在性较低的替代治疗药物。
食物的作用 :进食会使本品的生物利用度增加。本品不应与食物一起服用。服药前2小时之内和服药后1小时之内避免进食。任何时候都应该避免进食葡萄柚汁和其它已知的有抑制CYP3A4作用的食物。见【药物相互作用】。
对于不能吞下胶囊的患者,可以把胶囊的内容物与一茶匙的苹果酱混合在一起,混合后应立即服用。苹果酱不能超过一茶匙,同时不能食用除了苹果酱以外的其他食物。(见【用法用量】)。
肝损害 :肝损害对本品的药代动力学有轻度影响。与对照组中肝功能正常的受试者相比较,在轻度、中度或重度肝损害患者中,单剂量本品给药可导致AUC分别增长35%、35% 和19%。稳态下本品的预测Cmax分别增加了29%、18%和22%。临床研究中已经排除了ALT和/或AST> 2.5(或> 5,如果与疾病相关的话)倍正常值上限和/或总胆红素> 1.5倍正常值上限的患者。本品主要经肝代谢,因此,肝损害患者的暴露量可能增加,推荐在肝损害的患者中谨慎使用,并且应该密切监测这些患者的QT间期延长。
全胃切除:在全胃切除的患者中,本品的生物利用度可能会降低。在这类患者中应该考虑进行更为频繁的随访。
溶瘤综合征:本品治疗的患者中有发生溶瘤综合征的病例。大多数病例表现出疾病进展、高白细胞计数和/或脱水。由于可能发生溶瘤综合征(TLS),建议在开始本品治疗前,纠正有临床表现的脱水并治疗高尿酸血症。见【用法用量】。
乳糖 :本品含有乳糖,所以对于有遗传性半乳糖不耐受问题、严重的乳糖缺陷或葡萄糖-半乳糖吸收障碍的患者,不推荐使用本品。
实验室检查 :
对接受本品的患者,应该根据医生的判断进行一定频率的实验室检查。
1)血脂
在新诊断的CML患者中进行的III期研究中,接受尼洛替尼400 mg每日两次的患者中有1.1%出现3或4级胆固醇升高;然而,在接受300 mg每日两次的剂量组中未见3或4级胆固醇升高。建议在开始本品治疗前评估血脂,在治疗期间如出现任何临床指征时也需进行评估。由于一些降胆固醇药物会通过CYP3A4通路代谢,如果需要使用降脂药物,治疗开始前请参考【药物相互作用】。
2)血糖
在新诊断的CML患者中进行的III期研究中,接受尼洛替尼400mg每日两次的患者中有5.8%出现3或4级血糖升高;在接受300mg每日两次的剂量组中6.5%出现3或4级血糖升高。建议在开始本品治疗前评估血糖水平,在治疗期间如出现任何临床指征时也需进行监测。如果检查提示需要进行治疗,医生应根据当地实践和治疗指南进行治疗。
3)对驾驶能力和操作机器能力的影响:尚未进行过本品对驾驶能力和操作机器能力的影响的研究。不良反应中如头昏、恶心和呕吐,在本品治疗期间是有可能出现的,所以驾驶或操作机器时应该谨慎。
育龄期妇女
育龄期妇女在接受本品期间以及治疗结束后至少2周必须采取高效的避孕措施(可使怀孕率低于 1%的方法)。
妊娠
♦在服用本品期间,男性患者和有妊娠可能的女性患者必须采用有效的避孕措施。
♦除非必要,否则在妊娠期避免使用本品。
哺乳
♦由于该药对婴儿的风险尚不可排除,哺乳期妇女在服用达希纳期间和 最后一次给药后2周内不应该进行哺乳。
生育能力
尼洛替尼对于男性和女性生育能力的影响未知。在服用本品期间,男性患者必须采用有效的避孕措施。动物实验中,在雌性和雄性大鼠中给予最高试验剂量(大约高于人类推荐剂量的5倍)时,没有观察到药物对精子计数/活力及生育能力产生影响。
尚无在儿童或青少年中进行的临床研究。所以不推荐用于治疗小于18岁的患者。
对超过65岁的患者,不需要进行特殊的剂量调整。
本品是经肝脏中的CYP3A4代谢的,同时也是多重药物外排泵P-糖蛋白(Pgp)的底物。因此,本品的系统吸收(包括吸收及随后的消除)可能会受到影响CYP3A4和/或Pgp药物的影响。
1.可能增加本品血清浓度的药物
在一项I期研究中,尼洛替尼与伊马替尼(Pgp和CYP3A4的一种底物及调节剂)联合使用,两种药物都对CYP3A4和/或Pgp有轻微的抑制作用。将这两种药物合用时,伊马替尼的AUC从增加了18%至39%,尼洛替尼的AUC增加了18%至40%。
在健康受试者中,当与CYP3A4强抑制剂酮康唑合用时,本品的生物利用度增加了3倍。所以应该避免与酮康唑或其它强效CYP3A4抑制剂(如伊曲康唑、伏立康唑、克拉霉素、利托那韦、泰利霉素和其它蛋白酶抑制剂)同时使用。可以考虑没有或仅有弱的CYP3A4抑制作用的替代的合并用药。
2.可能减少本品血清浓度的药物
在接受CYP3A4诱导剂(利福平,每日600mg,持续给药12天)的健康受试者中,本品的系统暴露量(AUC)大约下降了80%。
CYP3A4活性诱导剂可提高尼洛替尼的代谢,从而降低尼洛替尼的血药浓度。同时服用CYP3A4诱导剂(如利福平、卡马西平、苯巴比妥、苯妥英和贯叶连翘)可能减少本品的暴露。在需要使用CYP3A4诱导剂的患者中,应该考虑具有较弱酶诱导作用的替代药物。
本品的溶解度具有pH依赖性,pH较高时溶解度较低。在接受埃索美拉唑(每日一次40mg,持续给药5天)的健康受试者中,胃pH值显着增加,但本品吸收只出现小幅下降(Cmax降低27%,AUC0-∞降低34%)。根据需要,本品可与埃索美拉唑或其他质子泵抑制剂同时服用。
在一项健康受试者研究中,法莫替丁给药后10小时和给药前2小时给予单剂量本品400 mg时,未观察到尼洛替尼的药代动力学出现显著变化。因此,当有需要合并使用H2受体阻滞剂时,可在本品给药前约10小时和给药后约2小时给予。
在同一项研究中,单剂量本品400 mg给药前后2小时给予一种抑酸剂(氢氧化铝和氢氧化镁/二甲基硅油)未改变尼洛替尼的药代动力学。因此,当有需要合并使用抑酸剂时,可在本品给药前后约2小时给予。
3.可能被本品改变血清浓度的药物
在体外,本品是CYP3A4、CYP2C8、CYP2C9、CYP2D6和UGT1A1的竞争性抑制剂,其中CYP2C9的Ki值最低(Ki=0.13 mM)。在健康受试者中,服用单剂本品和咪达唑仑,咪达唑仑的暴露增加了30%。当同时服用本品和这些酶的治疗指数窄的底物时,应该谨慎。在健康受试者中,临床相关浓度的尼洛替尼未改变华法林(一种敏感的CYP2C9底物)的药代动力学或药效动力学。在进行香豆素(CYP2C9 和CYP3A4的底物)治疗的患者中,应该增加对INR的监测。然而1-羟咪达唑仑与咪达唑仑的代谢比例不会改变。
同时给予经CYP3A4通路代谢的降胆固醇药物(如他汀类药物)时应谨慎,因为可能增加全身暴露(见【注意事项】)。
4.抗心律失常药和其它可能延长QT间期的药物
本品应该慎用于患有或可能发生QT间期延长的患者,包括服用抗心律失常药物,如服用胺碘酮、丙吡胺、普鲁卡因胺、奎尼丁、索他洛尔,或服用其它可能导致QT间期延长的药物,如氯喹、卤泛曲林(halofantrine)、克拉霉素、氟哌啶醇、美沙酮、莫西沙星、苄普地尔和匹莫齐特(见【注意事项】)。
5.与食物的相互作用
与食物一同摄入时,本品的吸收和生物利用度会增加,导致血药浓度升高。任何时候都应避免葡萄柚汁和其它抑制CYP3A4的食物。
已有药物过量的病例报告。有人将不明剂量的尼洛替尼胶囊与酒精和其他药物一起吞服,结果导致中性粒细胞减少、呕吐和困倦。当过量发生时,应密切观察患者并给予适当的支持治疗。
新诊断的Ph+ CML慢性期
研究CAMN107A2303全球数据
展开了一项开放性、多中心、随机 III 期研究(CAMN107A2303),目的是在通过细胞遗传学证实的新诊断 Ph+CML 慢性期成年患者中确定本品与伊马替尼相比的有效性。这些患者都在诊断六个月内,之前除羟基脲和/或阿那格雷外没有接受过 CML-CP 治疗。此外,在诊断时根据 Sokal 风险评分对患者进行了分层。
疗效评价以 846 例患者为基础,283 例在伊马替尼 400mg 每日一次组,282例在尼洛替尼 300mg 每日两次组,281 例在尼洛替尼 400mg 每日两次组。
三个组的基线特征均衡。在伊马替尼组的中位年龄为 46 岁,在两个尼洛替尼组中为 47 岁;在伊马替尼组、尼洛替尼 300mg 每日两次组和尼洛替尼 400mg 每日两次组中,分别有 12.4%、12.8%和 10.0%的患者≥65 岁。在所有组中,男性略多于女性(在伊马替尼组、尼洛替尼 300mg 每日两次组和尼洛替尼 400mg 每日两次组中,男性所占比例分别为 55.8%、56.0%和 62.3%)。60%以上的患者为白种人,25%为亚洲人。
所有 846 例患者完成(或提前终止)为期 12 个月的治疗时进行了主要数据分析。后续分析在患者完成 24,36,48 和 60 个月的治疗(或提前终止)时进行。在三个治疗组中,中位治疗持续时间大约为 60 个月。在伊马替尼组中,实际剂量强度中位数为 400mg/天,在尼洛替尼 300mg 每日两次组中为 593mg/天,在尼洛替尼 400mg 每日两次组中为 773mg/天。该项研究正在进行中。
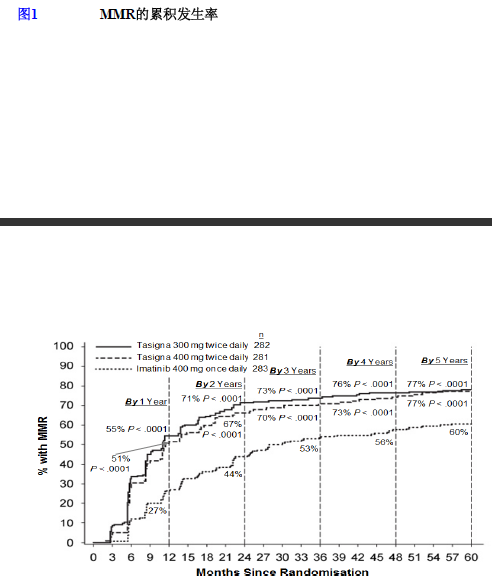
主要分子学反应(MMR)
主要疗效变量是指研究给药开始后 12 个月时的主要分子学反应(MMR)。主要分子学反应(MMR)被定义为以 RQ-PCR 测量的国际化量表为标准 BCR-ABL/ABL%≤0.1%,相当于 BCR-ABL 转录本水平从标准化基线降低≥3 个对数级。
与伊马替尼 400mg 每日一次组相比较,尼洛替尼 300mg 每日两次组中 12个月时的主要疗效终点,即主要分子学反应率显著较高(44.3% vs 22.3%,p<0.0001)。与伊马替尼 400mg 每日一次组相比较,尼洛替尼 400mg 每日两次组中12个月时的 MMR 率仍然较高(42.7% vs 22.3%,p<0.0001),见表 4。
在尼洛替尼推荐剂量 300mg 每日两次时,3 个月、6 个月、9 个月和 12 个月的 MMR 率分别为 8.9%、33.0%、43.3%和 44.3%。在尼洛替尼 400mg 每日两次组中,3 个月、6 个月、9 个月和 12 个月的 MMR 率分别为 5.0%、29.5%、38.1%和42.7%。在伊马替尼 400mg 每日一次组中,3 个月、6 个月、9 个月和 12 个月的 MMR 率分别为 0.7%、12.0%、18.0%和 22.3%。
12 个月、 24 个月、36 个月、48 个月和 60 个月的 MMR 率见表 5。

不同时间点的MMR率(包括在这些时间点或这些时间点之前获得MMR的患者均视为反应者)按MMR的累计发生率显示,见图1。
所有的Sokal风险分组中,所有时间点的MMR率对比结果均是两个尼洛替尼组高于伊马替尼组。
回顾分析发现,在治疗第 3 个月时,尼洛替尼 300mg 每日两次组有 91%(234/258)的患者的BCR-ABL水平≤10%,相比之下,伊马替尼400mg每日一次组有67%(176/264)的患者的BCR-ABL水平≤10%。在治疗第3个月时BCR-ABL水平≤10%的患者在第60个月的总生存率高于那些未达到这一分子学反应水平的患者(分别为97% vs. 82% [p=0.0116])。
基于至首次获得MMR的时间的Kaplan-Meier分析,在不同时间点获得MMR的患者比例在尼洛替尼300mg每日两次组和400mg每日两次组均高于伊马替尼400mg每日一次组(尼洛替尼300mg每日两次组vs.伊马替尼400mg每日一次组,HR=2.20,分层时序检验p<0.0001;尼洛替尼400mg每日两次组vs.伊马替尼400mg每日一次组,HR=1.90,分层时序检验p<0.0001)。
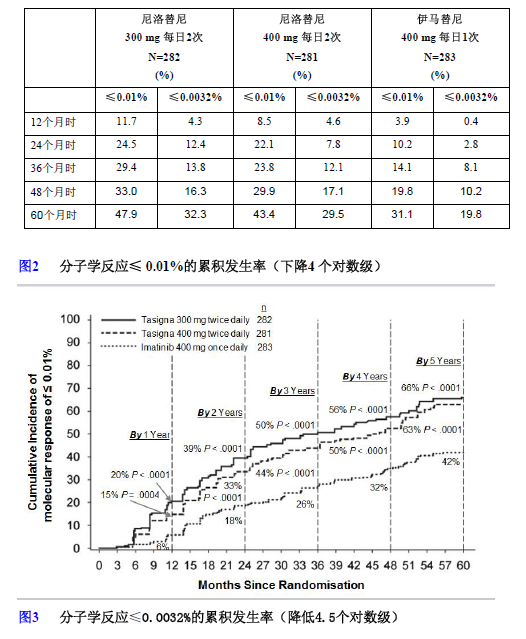
在不同时间点分子学反应(按国际单位制)达到≤0.01%和≤0.0032%的患者比例见表5,至不同时间点分子学反应(按国际单位制)达到≤0.01%和≤0.0032%的患者比例见图2和图3。BCR-ABL转录水平与标准化的基线水平相比,分子学反应(按国际单位制)达到≤0.01%和≤0.0032%分别相当于降低≥4个对数级和降低≥4.5个对数级。
表6 分子学反应≤0.01%(下降4个对数级)和≤0.0032%(下降4.5个对数级)的患者比例

MMR的持续时间
根据首次获得的MMR的持续时间Kaplan-Meier估算值,60个月后仍维持反
应的患者在获得MMR的患者中的占比在尼洛替尼300mg每日两次组为93.4%(95% CI:89.9% –96.9%)、尼洛替尼400mg每日两次组为92.40%(95% CI: 88.2% - 95.8%)和伊马替尼400mg每日一次组为89.1%(95%CI:84.2%-94.0%)。
完全细胞遗传学反应(CCyR)
完全细胞遗传学反应(CCyR)被定义为骨髓中Ph+中期分裂相细胞为0%(至少评估20个中期分裂相细胞)。至第12个月获得最佳CCyR(包括在第12个月或第12个月之前获得CCyR的患者均视为反应者)的患者比例在尼洛替尼300mg每日两次组和400mg每日两次组均显著高于伊马替尼400mg每日一次组,
见表7。
至第24个月获得CCyR(包括在第24个月或第24个月之前获得CCyR的患者均视为反应者)的患者比例在尼洛替尼300mg每日两次组和400mg每日两次组均显著高于伊马替尼400mg每日一次组。

CCyR的持续时间
基于Kaplan-Meier的估算值,获得CCyR并且60个月后仍维持反应的患者在获得CCyR的患者中的占比在尼洛替尼300mg每日两次组为99.1%(95% CI: 97.9-100%),在尼洛替尼400mg每日两次组为98.7%(95% CI: 97.1-100%),在伊马替尼400mg每日一次组为97.0%(95% CI: 94.7-99.4%)。
治疗中进展为AP/BC
治疗中进展至加速期(AP)或急变期(BC)定义为从随机化至首次证实疾病进展至加速期或急变期或CML相关的死亡的时间。观察到共有17例患者在治疗期间疾病进展为加速期或急变期:尼洛替尼300mg每日两次组2例,尼洛替尼400mg每日两次组3例,伊马替尼400mg每日一次组12例。出现病情进展的患者都没有达到MMR。然而,4例患者达到完全细胞遗传学反应(CCyR),这些患者都在伊马替尼组。在60个月时没有进展至加速期或急变期的估计患者比例分别为 99.3% 、 98.7% 和 95.2% (尼洛替尼 300mg 每日两次组 vs. 伊马替尼组的 HR=0.1599 ,时序检验 p=0.0059 ,尼洛替尼 400mg 每日两次 vs. 伊马替尼组HR=0.2457,时序检验p=0.0185)。自2年分析后,治疗中未见进展至加速期或急变期的新报告。
如果将克隆演变也作为疾病进展,截至数据截止日期,治疗期间疾病进展至加速期或急变期的患者共有25例(尼洛替尼300mg每日两次组中3例、尼洛替尼400mg 每日两次组5例、伊马替尼400mg每日一次组17例)。在第60个月时没有进展至加速期或急变期(包括克隆进展)的患者比例估计值分别为98.7%、97.9%和93.2%(尼洛替尼300mg每日两次组vs.伊马替尼400mg每日一次组,HR=0.1626,分层时序检验p=0.0009;尼洛替尼400mg每日两次组vs.伊马替尼400mg每日一次组,HR=0.2848,分层时序检验p=0.0085)。
总生存期(OS)
共有50例患者在治疗期间或是终止治疗后的随访期内死亡(尼洛替尼300mg每日两次组18例,尼洛替尼400mg每日两次组10例,伊马替尼400mg每日一次组22例)。这50例死亡中,26例死亡与CML有关(尼洛替尼300mg每日两次组6例,尼洛替尼400mg每日两次组4例,伊马替尼400mg每日一次组16例)。在第60个月时存活患者的估计比例分别为93.7%、96.2%和91.7%(尼洛替尼300mg每日两次组vs.伊马替尼400mg每日一次组,HR=0.8026,分层时序检验p=0.4881;尼洛替尼400mg每日两次组vs.伊马替尼400mg每日一次组,HR=0.4395,分层时序检验p=0.0266)。如果只考虑CML相关死亡事件,在第60个月时的估计总生存率分别为97.7%、98.5%和93.8%(尼洛替尼300mg每日两次组vs.伊马替尼400mg每日一次组,HR=0.3673,分层时序检验p=0.0292;尼洛替尼400mg每日两次组vs.伊马替尼400mg每日一次组,HR=0.2411,分层时序检验p=0.0057)
伊马替尼治疗后未取得大于或等于4.5个对数级下降的分子学反应的Ph+CML-CP成年患者换用尼洛替尼
在一项开放性、多中心、随机、III期研究中,入组了207名接受伊马替尼治疗至少两年的Ph+ CML-CP成年患者,这些患者在进入研究之前的6个月内未进行永久性的伊马替尼剂量调整,并且在进入研究之前的3个月内没有出现主要毒性。按1:1的比例将患者随机分配至接受本品400 mg每日两次给药(n=104)或者继续接受与随机前相同剂量(400 mg或600 mg,每日一次)的伊马替尼治疗(n=103)。按照先前伊马替尼治疗的持续时间以及先前干扰素使用的持续时间对随机分组患者进行分层。在截止日期时,尼洛替尼治疗组、伊马替尼400mg和600mg治疗队列的中位治疗时间(从治疗的第一天到随机化治疗的最后一天)分别为47.2个月、37.0个月和26.7个月。两个治疗组的人口统计学和基线特征均衡(包括入组时的BCR-ABL转录本水平)。尼洛替尼组和伊马替尼组患者的中位年龄分别为46岁和52岁,这两组分别有13.5%和13.6%的患者年龄≥65岁。男性患者多于女性患者(尼洛替尼组和伊马替尼组的男性患者比例分别为68.3%和63.1%)。超过80%的患者为白种人。至截止日期为止,尼洛替尼治疗组的中位实际剂量强度为775.7 mg/天,伊马替尼治疗组两个剂量队列则分别为400 mg/天和600 mg/天。
该研究的主要终点为尼洛替尼或伊马替尼治疗1年内确认最佳累积完全分子学反应(CMR)率。在第一个12个月期间,尼洛替尼组和伊马替尼组确认最佳累积CMR率分别为12.5%和5.8%。在头12个月时间点时,主要终点没有统计学意义(p=0.1083),比值比(OR)为2.096,对尼洛替尼有利。
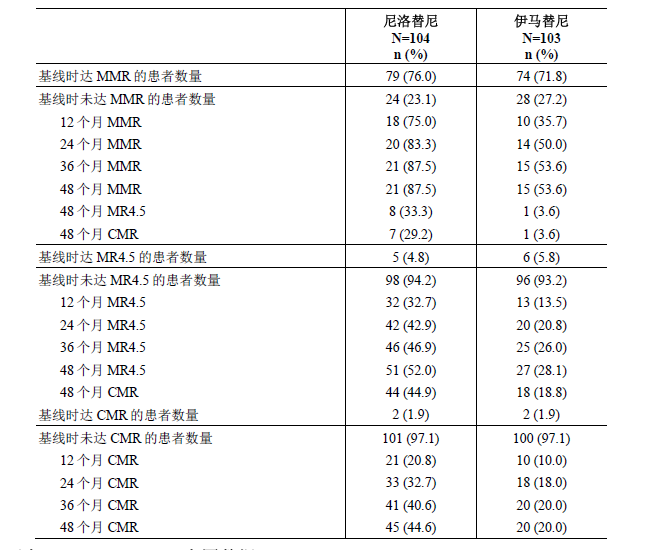
48个月的主要结局变量长期随访为次要终点。评估基线时无相应反应患者转换治疗后所取得的不同分子学反应水平的分析表明,从伊马替尼转换至尼洛替尼之后,在第48个月时,随机治疗下取得MMR、MR4.5和CMR的患者数量会出现有临床意义的升高(见表8)。 表8 按基线分子学反应状态总结转换治疗后取得最佳累积分子学反应的患者比例
研究CAMN107ECN02中国数据
CAMN107ECN02为尼洛替尼在在新诊断的、既往未治疗过的Ph+ CML-CP中国成人患者中进行的开放性、多中心、随机III期注册研究,研究目的为比较伊马替尼400mg每日一次与尼洛替尼300mg每日两次的有效性。
研究共入选了267例患者,患者在诊断6个月内,除了羟基脲、阿那格雷和不超过3个月的干扰素或不超过14天的伊马替尼,患者既往未接受过Ph+ CML-CP的其他治疗。按照Sokal评分和既往是否接受了干扰素(IFN)治疗进行分层随机,133名患者随机分入伊马替尼400mg每日一次组,134名患者随机分入尼洛替尼300mg每日两次组。允许在疗效不佳或治疗失败的情况下进行伊马替尼组(400mg/天到600mg/天)的剂量递增(优化)。尼洛替尼组不允许剂量增加。
两组的基线特征均衡。伊马替尼组的中位年龄为39岁,尼洛替尼组的中位年龄为41岁,伊马替尼组和尼洛替尼组分别有3.0%和5.2%的患者年龄≥65岁。
两治疗组均为男性患者多于女性患者(伊马替尼和尼洛替尼组分别为60.9%和67.9%)。整体上,16%的患者为Solak高风险,52%的患者为Sokal低风险。
所有267例患者完成约12个月(12个周期,每周期28天)治疗或提前退出研究时进行主要数据分析。至分析的数据截止日,两组的中位治疗持续时间均为11.6个月。伊马替尼400mg每日一次组的中位实际剂量强度为392.7mg/日,尼洛替尼300mg每日两次组为565.2mg/日。
主要分子学反应(MMR)
研究的主要终点是治疗12个月后的MMR率。MMR定义为在开始首次研究药物治疗后12个月时通过以RQ-PCR测得的国际化量表为标准的BCR-ABL/ABL≤0.1%。
在12个月时,尼洛替尼300mg每日两次组的MMR率较伊马替尼400mg每日一次组显著更高(52.2% vs 27.8%,p<0.0001)高。尼洛替尼300mg每日两次组在3个月、6个月、9个月时的MMR率分别为13.4%,44.8%和47.8%,伊马替尼400mg每日一次组在3个月、6个月、9个月时的MMR率分别为3.0%,18.0%和21.1%。在不同sokal评分的患者中,尼洛替尼300mg每日两次组的MMR率也均高于伊马替尼400mg每日一次组,详见表9。
表9 各Sokal风险组12个月时的MMR率

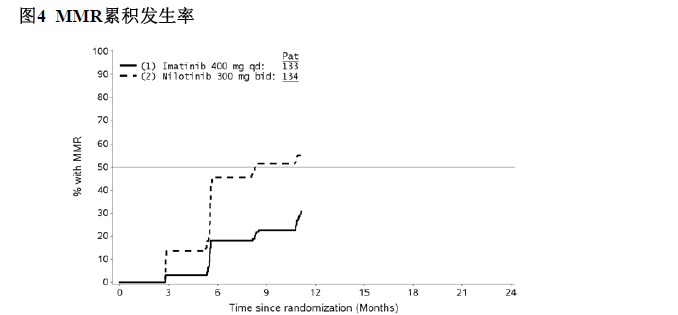
12个月累积MMR发生率为以Kaplan-Meier(K-M)分析标示以在12个月时或之前获得MMR的患者(如图4),伊马替尼400mg每日一次组的累积MMR发生率为30.8%,尼洛替尼300mg每日两次组累积MMR发生率为56.0%。伊马替尼组至首次MMR的平均时间为7.10 个月,尼洛替尼组为 5.59个月。不同时间点时达到 MMR 的概率在尼洛替尼组中高于伊马替尼组 ( 尼洛替尼和伊马替尼之间HR=2.4218,分层时序检验p<0.0001)。
完全细胞遗传学反应(CCyR)
CCyR定义为分析至少20个骨髓分裂中期细胞Ph+细胞比例为0%,12个月时最佳CCyR率将在12个月时间点时或之前达到CCyR的患者作为有疗效者。尼洛替尼300mg 每日两次组的 6 个月时最佳 CCyR 率(66.4%)高于伊马替尼组(57.1%)。在12个月时两个治疗组的最佳CCyR率均为77%。
疾病进展、事件和总生存
不将克隆演变计为进展,每个治疗组至截止日期每个治疗组有2名患者进展至AP/BC,按Kaplan-Meier(K-M)法分析,伊马替尼400mg每日一次组和尼洛替尼300mg每日两次组12个月时未进展至AP/BC患者的估计比例分别为98.5%和98.4%。此外,每个治疗组有1名患者证实丧失CCyR,两组中12个月的估计无事件生存率均为97.7%。
在将克隆演变患者也考虑为进展至AP/BC时,至截止日伊马替尼组另1名患者和尼洛替尼组另2名患者考虑为进展至AP/BC。12个月时未进展至AP/BC患者的估计比例分别为96.8%和96.9%。
伊马替尼组中1名患者和尼洛替尼组中2名患者死亡。所有死亡均发生于停药28天后的生存随访期,其中仅1例发生在尼洛替尼组的死亡为CML相关死亡。各治疗组中12个月时存活患者的估计百分率分别为99.2%和98.5%。
安全性
尼洛替尼组的总体安全性与伊马替尼组相当。尼洛替尼组发生率≥10%的不良事件为皮疹、血小板减少症、血胆红素升高、脂肪酶升高、高胆红素血症、白细胞减少症、ALT升高、血小板计数减少、肌痛、白细胞计数减少和中性粒细胞计数减少。这些不良事件大多为1级和2级,其中发生率≥5%的3级或4级不良事件为血小板减少症(20.3% )、脂肪酶升高(12.0% )、白细胞减少症(6.0%)和中性粒细胞计数减少(10.5%),这些事件通常在短暂停药后缓解。4名患者(3.0%)发生QT间期延长>450毫秒,未见QT间期延长>480毫秒,也无尖端扭转综合症和及猝死病例。1名患者发生因血小板减少导致的脑出血,















