
利鲁唑片
Riluzole Tablets
Li Lu Zuo Pian
本品主要成份为利鲁唑
化学名称:2-氨基-6-三氟甲氧基苯并噻唑
化学结构式: 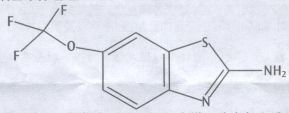
分子式:C8H5F3N2OS
分子量:234.20
本品为白色薄膜衣片,除去薄膜衣片后显类白色。
用于肌萎缩侧索硬化症。
50mg
口服,一次一片,(50mg),一日2次。增加每日剂量不会增加药效,但增加不良反应。如漏服一次,按原计划服用下一片。
餐前或餐后两小时服药,以降低食物对利鲁唑生物利用度的影响。
在利鲁唑用于ALS患者的Ⅲ期临床研究中,最常见报告的不良反应为乏力、恶心和肝功能检测异常。
不良反应按照其发生率排序列出如下,使用如下约定:非常常见(≥1/10),常见(≥1/100至<1>血液和淋巴系统异常 不常见:贫血
未知:严重中性粒细胞减少症(见【注意事项】)
免疫系统异常 不常见:类过敏反应、血管性水肿
神经系统异常 常见:头疼、眩晕、感觉异常、嗜睡
呼吸、胸部和纵隔疾病 不常见:间质性肺病 (见【注意事项】)
心脏异常 常见:心动过速
胃肠道异常 非常常见:恶心
常见:腹泻、腹痛、呕吐
不常见:胰腺炎
肝胆-异常 非常常见:肝功能检测异常。丙氨酸氨基转移酶的增高通常发生于利鲁唑治疗的前3个月内 ;其通常为一过性.且当治疗继续时,其水平在2至6个月内恢复至低于正常上限2倍。这些増 高可伴有黄疸。在临床试验中ALT升高超过正常范围上限5倍的患者中止治疗后ALT水平在2~ 4个月内恢复至正常范围上限2倍以下。(见【注意事项】)
未知:肝炎
一般性异常及纶药部位状况 非常常见:乏力
常见:疼痛
-对本品及其主要成份过敏者。肝功能不正常或转氨酶水平异常升高者,处于妊娠及哺乳期禁用。
肝损害:
利鲁唑慎用于有肝功能异常史的患者,或血清转氨酶(ALT/SGPT; AST/SG0T升至正常上限3倍)、胆红棄和/或γ-谷氨酰转移酶(GGT)水平轻度增高的患者。肝功能检测的基线堆高(特别是胆红素升高)须禁止利鲁唑的使用(见【不良反应】)。
因为有肝炎的风险,在利鲁唑治疗前和治疗过程中应该进行血清转氨酶,包括ALT的检测。在治疗最初3个月,须每月检测ALT,在第1年每3个月检测1次,以后每年一次。在发生ALT水平增高的患者,须进行更为频繁的ALT水平的检测。
如果ALT水平堆加至5倍ULN,利鲁唑须停药。在发生ALT増加至5倍ULN的患者尚无减量或再次给药的经验。不推荐利鲁唑在这种情况患者的再次给药。
中性粒细胞减少症:
须警告患者向其医生报告所有的发热疾病。发热疾病的报告须提醒医生检查白细胞计数,在中性粒细胞减少情况下停止利鲁唑的使用(见【不良反应】)。
间质性肺病
已有接受利鲁唑治疗报告间质性肺病的病例,其中一部分病例为严重病例(见【不良反应】)。如果出现呼吸症状,例如干咳和/或呼吸困难,应进行胸部X线检查,如果有提示间质性肺炎的发现(例如两侧肺弥散不透明),应立即停用利鲁唑。在大部分报告的病例中,停药和对症治疗后,症状消除。
肾损害:
在中度或重度慢性肾功能不全(肌酐清除率在10-50ml/min)的患者和健康志愿者单次口服50mg利鲁唑给药后,其药代动力学参数无显著差异。本品不推荐用于肾功能损害的患者,因为在此人群尚未进行重复给药的研究。
对驾车和使用机器能力的影响
须警告患者有头晕或眩晕的可能,并建议其当发生这些症状时不要驾车或操作机器。
尚无对驾车和使用机器影响的研究。
孕期和哺乳期的妇女禁用。
尚未明确。
老年患者用药同健康成人。
在孤立病例曾观察到神经和精神症状,急性中毒性脑病伴木僵、昏迷,以及高铁血红蛋白血症。
在过量病例,行对症和支持治疗。
采用亚甲蓝治疗后可迅速逆转重度高铁血红蛋白血症。
肌萎缩侧索硬化症(ALS)的发病机理尚未完全阐明,有学说认为谷氛酸在此疾病中是造成细胞死亡的原因之一。 利鲁唑的作用机制尚不清楚,其作用可能与抑制谷氨酸释放、稳定电压依赖性钠通道的失活状态、干扰神经递质与兴奋性氨基酸受体结合后细胞内事件有关。一项动物实验显示,利鲁唑能延长ALS转基因动物模型的存活时间。多种神经兴奋性损伤动物模型研究显示利鲁唑具有神经保护作用。体外研究显示,利鲁唑能保护培养的大鼠运动神经元免受谷氨酸的兴奋性毒性损伤,并抑制缺氧引起的皮层细胞死亡。
毒理研究:
遗传毒性
利鲁唑Ames试验、L5178Y细胞基因突变试验、大鼠细胞遗传学试验、小鼠微核试验结果均为阴性;人淋巴细胞染色体畸变试验结果意义不明确,第二次试验未能重复该结果。
利鲁唑主要活性代谢产物N-轻基利鲁唑小鼠淋巴瘤试验和体外L5178Y细胞微核试验结果阳性,L5178Y细胞HPRT基因突变试验、Ames试验、大鼠肝细胞UDS试验、人淋巴细胞染色体畸变试验、小鼠微核试验结果均为阴性。
生殖毒性
雄性和雌性大鼠经口给予利鲁挫15mg/kg(按mg/m2推算,约相当于人日最大给药剂量的1.5倍),未见对生育力的影响。大鼠和家兔致畸敏感期分别经口给予利鲁唑27mg/kg和60mg/kg(按mg/m2推算,分别约相当于人日最大给药剂量的2.6和11.5倍),可见母体毒性。雄性和雌性大鼠自交配前至围产期经口给予利鲁挫15mg/kg(按mg/m2推算,约相当于人日最大给药剂量的1.5倍),可见着床率降低、胚胎死亡增加,幼仔存活力降低,生长减慢。
致癌性
小鼠和大鼠分别经口给予利鲁唑20mg/kg和10mg/kg(按mg/m2推算,约相当于人日最大给药剂置)连续2年, 未见致癌性。
在健康男性志愿者中,通过单一剂量口服25至300 mg以及每日两次重复口服25至100mg对利鲁唑的药代动力学进行评估。其血药浓度水平的升高与剂量呈线性关系,其药代动力学特性是非剂量依赖性的。
重复剂量给药时(50 mg利鲁唑片,每日两次,十天疗程),利鲁唑原形在血浆中蓄积至单一剂量的2倍,并于5日内达到稳态期。
吸收:利鲁唑口服后吸收迅速,并于60至90分钟内达最大血浆浓度(Cmax=173±72(sd)ng/ml)。大约剂量的90%被吸收,绝对生物利用度为60±18%。
在高脂饮食的同时服用利鲁唑,其吸收率及吸收程度下降。(Cmax降低44%,曲线下面积降低17%)。
分布:利鲁唑在体内分布广泛,可通过血脑屏障。利鲁唑的分布容积大约为245±69升(3.4升/公斤体重)。
利鲁唑的蛋白结合率大约为97%,主要与血浆白蛋白及脂蛋白结合。
代谢:利鲁唑主要以原形存在于血浆中,并由细胞色素P450广泛代谢继而糖脂化。在体外试验中利用预备的人体肝脏显示细胞色素P450 1A2为主要的利鲁唑代谢有关的同功酶。在尿中的代谢产物为3种酚衍生物,1种脲基衍生物及原形利鲁唑。
已鉴别和非结合的代谢产物在动物中不显示利鲁唑的药效特性,因此在人体中未做研究。
排泄:排泄半衰期范围在9至15小时。利鲁唑主要从尿液中排出。
尿中总排泄率为剂量的90%。葡萄糖醛酸衍生物占尿中代谢产物的85%以上。仅有剂量2%的利鲁唑以原形存在于尿中。
特殊人群
老年人:在老年人(>70岁)中利鲁唑多次口服给药(50毫克利鲁唑每天两次治疗4.5天)的药代动力学参数不受影响。
肝损伤:利鲁唑50毫克单次口服给药后,在轻度慢性肝功能不全的患者中,AUC大约升高1.7倍,在中度慢性肝功能不全的患者中,大约升高3倍。
肾损伤:在中度或重度慢性肾功能不全(肌酐清除率在10和50毫升/分钟之间)的患者和健康志愿者中利鲁唑50毫克单次口服给药后,药代动力学参数没有明显的差异。
人种:在健康的日本和高加索成年男性中进行了一项临床研究,每天两次给药8天后,评价利鲁唑及其代谢产物 N-羟基利鲁唑的药代动力学。在日本和高加索受试者中,没有发现利鲁唑及其代谢产物的药代动力学参数有人种间的差异。
遮光,密封保存。
铝塑包装,12片/板×2板/盒。
暂定24个月
进口药品注册标准:JX20090316
国药准字H20073149
FDA妊娠分级:C















